Noonan-Syndrom: Beschreibung, Symptome, Behandlung
Es gibt eine Vielzahl von Geburtsschäden bei Kindern. Einige von ihnen werden als durchaus üblich sein. Aber es ist eine seltene Erkrankung. Ihre Prävalenz ist niedrig. Doch viele sind sehr schwerwiegende Folgen haben. Eine dieser Erkrankungen ist Noonan-Syndrom. Als nächstes wollen wir uns es genauer betrachten. Dieser Artikel beschreibt, wie und warum es erscheint Noonan-Syndrom. Foto Krankheit wird auch im Text dargestellt werden. 
Überblick
Noonan-Syndrom ist eine erbliche Pathologie. PTPN11 Gen mutiert durch das Tragen seiner Eltern auf die Nachkommen übertragen. In der Regel Männer in dieser fruchtlos. Daher wird das Gen durch die mütterliche Linie übertragen. In der Regel Familiencharakter der Krankheit gekennzeichnet. Seltene Fälle von nicht-erblich, aber sie sind auch in der Praxis aufgenommen.
historische Informationen
Zu einer Zeit als Kardiologe praktiziert, Kinderarzt Zhaklin Nunan, in der Klinik der University of Iowa arbeitete, bemerkte ich, dass eine bestimmte Gruppe von Kindern, die von Jungen und Mädchen bestanden, die Pulmonalstenose hatte, oft durch geringes Wachstum gekennzeichnet, voneinander weit aufgerissenen Augen, webbed Hals niedrige Vorhöfe und ptosis entfernt. Die Untersuchung der Kombination von Manifestationen von Herzerkrankungen mit anderen Entwicklungsanomalien bei 833 Patienten, schrieb der Arzt in einem 1962 Artikel. In der Arbeit beschrieb sie neun Fälle , in denen auf dem Hintergrund der Herzfehler angeborene Art und charakteristische Gesichtszüge, Brust Verformung und Verkümmerung. Die Krankheit wurde in Menschen nachgewiesen männlich und weiblich. 
Noonan-Syndrom: Symptome
Es gibt eine Reihe von Merkmalen, die von anderer Pathologie unterscheiden. Dazu gehören insbesondere:
- Geringes Wachstum. Frauen – 1,53, für Männer -. 1,63 m zum Zeitpunkt der Geburt als die Körperlänge und Gewicht des Kindes ist innerhalb der normalen Grenzen. Verkümmerung beginnt ab dem Alter von 2-3 Jahren.
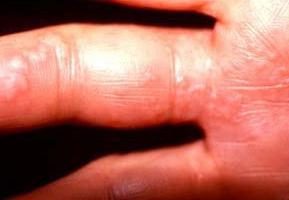
- Änderungen gegenüber. Patienten diagnostiziert Noonan – Syndrom, haben weit auseinander liegenden Augen Mandelform. In der inneren Ecke der Hautfalte vorhanden ist. Ebenfalls festgestellt Ptosis (Augenlid weggelassen) aufgrund gestörter Muskelfunktion, entweder wegen der kleinen Steckdosen. Etwa zwei Drittel aller Kinder mit Erkrankungen sind Störungen des Sehvermögens. Wenn Ptosis begrenzte Sichtfeld und entwickelt ein Schielen.
- Gebäude Verletzungen Backen. Oberunterentwickelt, ist es gebogener hohe Himmel. Beide Backen mit Zähnen Zwischenschaltung falsch.
- Flaches oder Deformation der Ohren. Als Ergebnis dieser Anomalie sind hörgeschädigt.
- Kurz und breit Hals.
- Schildförmigen Kasten mit weit auseinander liegenden Brustwarzen.
- Deformation des Ellbogens (kongenitale).
- Plattfuß.
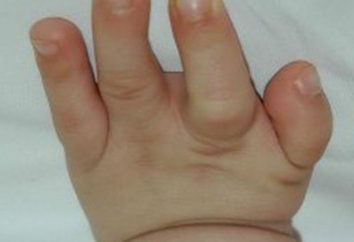
- Kurze Finger.

Fehlbildungen der inneren Organe
Noonan-Syndrom wird oft durch Störungen im Herz-Kreislauf-System begleitet. Vor dem Hintergrund der Pathologie ergab (Stenose) im Lungenstamm und den Defekt in der Scheidewand verengt. Der zweite Platz wird durch Anomalien des Urogenitalsystems besetzt. So zeigten die Nieren Defekte wie Hypoplasie (fehlende Gewebe) oder das Fehlen einer Niere. Hinsichtlich der Pubertät, es variiert von normal vollständig defekt. Mädchen oft nach dem Einsetzen der Menstruation beobachtet, bei Jungen – Hoden oder Kryptorchismus sind völlig fehlen. Auch stellte eine Verletzung der Spermatogenese. In einigen Fällen sind die Spermien völlig fehlen. Vor dem Hintergrund der Operationen für Noonan-Syndrom haben ein erhöhtes Blutungs. Einige Patienten können auch Anomalien zeigen , der geistigen Entwicklung in der mild. 
Form
Es gibt zwei Arten von Pathologie:
- Familiäre Form. Es unterscheidet sich erblich autosomal-dominant Typ. In Träger des mutierten Gens Nachkommen scheinen mit der Pathologie.
- Die sporadische Form. In diesem Fall scheint die Mutation von Fall zu Fall. In dieser Erbanlage wird nicht offenbart.
Gründe
Die häufigste Faktor, der die Pathologie provoziert ist die PTPN11-Genmutation. Diese Ursache ist in 50% der Patienten diagnostiziert. Allerdings zu einem bestimmten Prozentsatz der Menschen mit diesem Syndrom ist ein genetischer Faktor ist unklar. Vererbung Pathologie tritt in autosomal-dominant. Das Syndrom kann durch eine neue Mutation ausgelöst werden. Daraus folgt, dass die Eltern mit dem Gen haben nicht viel von einer Chance, ein weiteres Kind zu haben.
Diagnose
Es wird in Übereinstimmung mit den charakteristischen Merkmalen externen montiert (oben beschrieben). Auch in den Diagnose studierte Laborparameter. Insbesondere gibt es eine Abnahme der Testosteronkonzentration, Gerinnungsfaktor XII. Darüber hinaus gibt es Tools und Forschung. Ernennung durch die Röntgen des Brustbeins, Echokardiographie, EKG. mit diesen Techniken ergab Anomalien der inneren Organe. Der Arzt kann auch Beratung vorschreiben der medizinischen Genetik. 
therapeutische Maßnahmen
Im Zusammenhang mit der Behandlung genetisch bestimmt Syndrom richtet sich in erster Linie auf die Symptome beseitigt. Wenn eine Operation zugewiesen cryptorchidism. Während ihrer Hoden bewegen in den Hodensack. Mit der Reduzierung der Konzentration von Androgen-Hormon-Therapie wird empfohlen. In Anwesenheit von Nierenversagen, ist die Hämodialyse ernannt. Mit ihm wird die Entfernung aus dem Körper über Stoffwechselprodukte extrarenalen Blutreinigung durchgeführt.
Latest posts










Was ist Hepatitis C?
Gesundheit

